
本发明涉及生物高通量数据分析技术领域,具体涉及从动植物转录组数据中挖掘内生微生物组信息的方法。
背景技术:
微生物在自然界几乎无处不在,例如河流、湖泊、海洋、土壤、空气、人体表面以及人类、动植物的内部。许多微生物包括细菌,古细菌和真菌等都可以生活在植物的组织中。内生菌是一种以共生或有益的方式存在于宿主植物的各种组织中或不会引起任何有害作用的微生物。少数内生菌可以增强宿主植物对非生物胁迫(例如耐热性)的耐受性,而其他内生菌则可以通过产生植物激素、溶解磷和钾、生物固氮、抑制乙烯的生物合成来促进植物的生长。另外,许多内生菌可以通过产生氨、铁载体等方式来保护植物免受微生物病原体的侵害。动物以及人体组织中也发现很多内生微生物,比如癌症组织。
现有的内生菌检测方法主要包括:(1)分离培养法检测;(2)非培养法检测。非培养法主要为:利用pcr扩增特定细菌或者真菌共有区域后结合第二代或者第三代测序技术测序,比如扩增16srrna、真菌its区域或扩增特定目的基因后测序;直接对植物组织全基因组dna测序后,结合微生物组分析工具加以分析。利用扩增子分析,或者动植物组织全基因组测序均需要单独进行实验操作,成本较高。
技术实现要素:
为了解决上述问题,本发明提供了一种全新的对动植物内生菌进行研究的方法,从动植物转录组数据中挖掘内生微生物组信息的方法。
本发明通过以下技术方案来实现上述目的:
从动植物转录组数据中挖掘内生微生物组信息的方法,步骤包括:
(1)对宿主动植物转录组测序原始数据进行清理,以获得仅含高质量序列的转录组数据;
(2)将所述转录组数据与宿主参考基因组进行比对,获得含有微生物序列的数据文件;
(3)通过分析所述数据文件获得内生微生物的组成信息。
进一步改进在于,步骤(1)中,利用清理软件对宿主动植物转录组测序原始数据进行清理。
进一步改进在于,所述清理软件为trimmomatic。
进一步改进在于,步骤(2)的具体操作为:使用转录组比对软件将转录组数据与宿主动植物基因组或者全部cdna数据文件进行比对,同时区分输出比对上的植物编码基因转录组数据和未比对上的数据文件,利用其输出未比对上序列参数“--un-conc”获得未比对上的数据文件,所述数据文件中包含有一部分宿主动植物序列外,还包括表达的微生物基因片段。
进一步改进在于,所述转录组比对软件为hisat2软件或bowtie2软件。
进一步改进在于,步骤(3)中,利用微生物组解析软件,对未比对上的数据文件进行分析,结合bracken挖掘出植物内生微生物组成信息。
进一步改进在于,所述微生物组解析软件为kraken2。
本发明的有益效果在于:本方法与利用扩增子分析或者动植物组织全基因组测序分析微生物组成信息相比,不但可以分析出各类微生物组成及含量信息,还得到以往的宿主动植物基因表达信息,节约实验研究成本。
附图说明
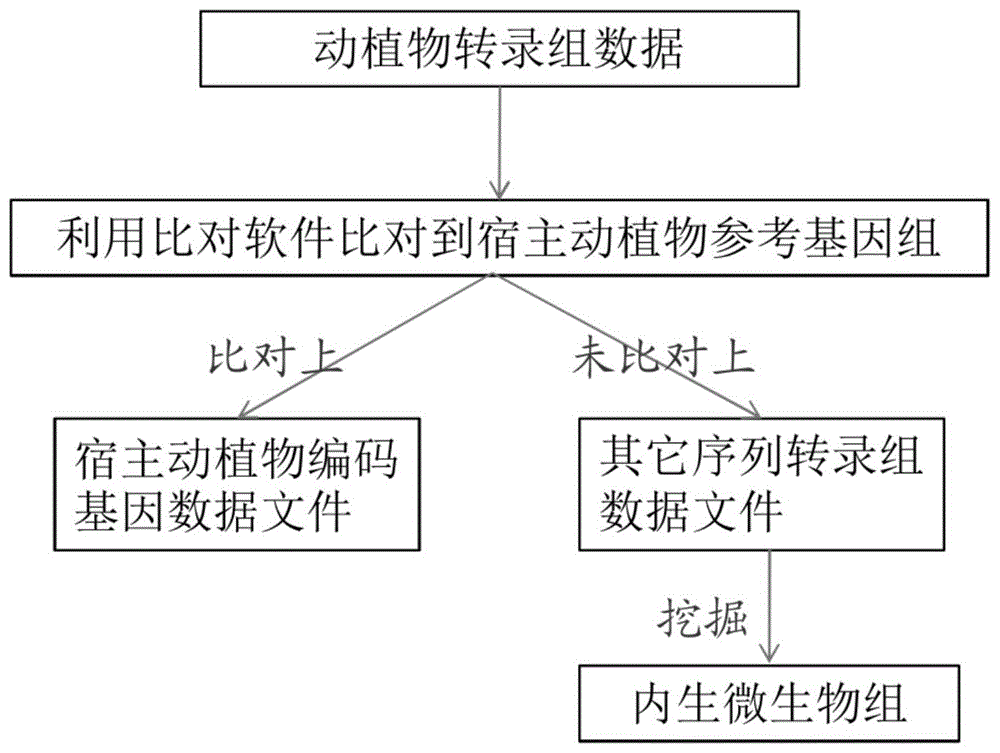
图1为本发明的技术方案路线图;
图2为根内生菌物种组成情况,图中,a为桑基流动图;b为内生菌组成情况结果图。
具体实施方式
下面结合附图对本申请作进一步详细描述,有必要在此指出的是,以下具体实施方式只用于对本申请进行进一步的说明,不能理解为对本申请保护范围的限制,该领域的技术人员可以根据上述申请内容对本申请作出一些非本质的改进和调整。
实施例1
本实施例提供了一种从动植物转录组数据中挖掘内生微生物组信息的方法,如图1所示,步骤包括:
(1)宿主动植物转录组测序原始数据清理:使用开源软件trimmomatic,使用默认参数:java-jartrimmomatic-0.33.jarpeinput_forward.fq.gzinput_reverse.fq.gzoutput_forward_paired.fq.gzoutput_forward_unpaired.fq.gzoutput_reverse_paired.fq.gzoutput_reverse_unpaired.fq.gzilluminaclip:truseq3-pe.fa:2:30:10leading:3trailing:3slidingwindow:4:15minlen:36
(2)获得含有微生物序列的转录组数据文件
利用开源软件hisat2-build建立宿主动植物参考基因组索引文件,参考命令:hisat2-buildgenome.fageome_index。利用hisat2将转录组测序文件与宿主动植物参考基因组索引文件进行对比,在分析基因表达的同时,输出未比对上的文件。参考命令:hisat2-xgeome_index-1forward.fq-2reverse.fq-p32--un-concun-conc_$sample_name.fastq-s$sample_name.sam2>$sample_name.log。
(3)解析微生物组成
步骤1.使用以下命令将序列数据文件与建立的包括细菌、真菌、病毒、动植物基因组的数据文件进行对比。参考命令:
catclean_reads_without_host_dna/*1.fastq>all_reads.1.fastq
catclean_reads_without_host_dna/*2.fastq>all_reads.2.fastq
kraken2--db=/ncbi_nr_nt_data/db_for_kraken2/kraken2--threads=24--pairedall_reads.1.fastqall_reads.2.fastq>database.kraken
步骤2:计算输入文件序列中每个完美的序列并进行分类。参考命令:
./kmer2read_distr--seqid2taxid${kraken_db}/seqid2taxid.map--taxonomy${kraken_db}/taxonomy--krakendatabase.kraken--outputdatabase${read_len}mers.kraken-k${kmer_len}-l${read_len}-t${threads}
步骤3:生成kmer分布文件。参考命令:
generate_kmer_distribution.py-idatabase${read_len}mers.kraken;-odatabase${read_len}mers.kmer_distrib
步骤4:产生微生物分类文件和报告文件。参考命令:
#kraken2--db=${kraken2_db}--threads${threads}--report${sample}.kreport2${sample}>${sample}.kraken2。
以玉米根转录组数据为例,从图2a可以看出,除玉米外,转录组数据中还包括真菌、细菌、古细菌、病毒,进一步可以获得各类微生物之间的含量关系(图2b)。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。

